No time to read now?
-> Download the article as a handy pdf
List of contents
EMA report echoes our calls on MAHs to stop signal detection activities in EudraVigilance
Analysis of the EMA’s pilot program report
Martti Ahtola & Dominik Hodbod | May 17, 2022

The text below is a longer read with a deeper analysis of the report and its meaning. For us, we’re even more committed in our quest to modernize pharmacovigilance and bring it to what can be called as value-based healthcare. It’s time to stop performing meaningless, time-consuming and expensive activities without any legal justification that only benefit manual labor-focused service providers who are on the income side of the invoice.
Summary
- The report, previously publicly unavailable, gives us confidence in our approach and calls on EU MA Holders to stop performing needless signal detection activities, as well as lobby with legislators to take action.
- Now that we have it, we will dig into the report’s key points. To us, its contents were both expected and surprising.
- We were surprised by the low amount of signal notifications from the MAHs because we assumed there to be a larger number of false and/or duplicate notifications.
- Most surprising part was that EMA admitted that there was no evidence to support the requirement of marketing authorization holders (MAH) monitoring of EudraVigilance for signal detection and that the best solution would be to remove the legal requirement instead of extending the process beyond the pilot companies.
- Among the expected, there were things such as the low quality of the signal notifications and the low amount of useful data gathered in the program in general.
Preface of the pilot
The use of EudraVigilance by the pharmaceutical companies for signal detection was introduced as a new feature with the aim to complement an established process where EudraVigilance data was monitored by EMA and the National Competent Authorities (NCA). One of the potential risks EMA expected beforehand was that a high volume of signal notifications from pharmaceutical companies could be received and the cases would have to be processed by EMA and NCAs within tight legal timelines. EMA’s assessment was that this would require diverting resources from pharmacovigilance activities with proven public health impact to signal review activities that have minor or no shown impact on public health. To avoid potential risk to public health, EMA and the European Commission agreed to a phased implementation of the legal requirements while gaining experience with the new process. First phase being the pilot program. Pharmaceutical companies included in the pilot, along with EMA and local regulatory agencies, were required to monitor EudraVigilance data using a risk-proportionate frequency, considering the profile of a given active substance or medicinal product. According to the pilot plan, each organization should determine the appropriate monitoring periodicity for each active substance / medicinal product, but EMA highlighted that the Guideline on good pharmacovigilance practices (GVP): IX – Signal Management recommends monitoring EV data at least every 6 months. A more frequent monitoring was advised for active substances contained in medicinal products subject to additional monitoring unless the sole reason for the additional monitoring status is the request for a post-authorization safety study (PASS).The results of the pilot
The report from EMA to European Commission states that while the direct involvement of the pharmaceutical companies in EudraVigilance monitoring may strengthen the EU signal management system with the detection of more safety signals, it may also be associated with some risks. The potential risks would be mainly duplication of signal detection activities between EMA, local regulatory authorities, and the pharmaceutical companies, as well as amongst pharmaceutical companies with medicinal products containing the same active substance.
EudraVigilance data is currently monitored at substance level, so the pharmaceutical companies receive data also for other companies with the same active pharmaceutical ingredient. For example, if the pharmaceutical company has a product with paracetamol as the active substance, in addition to their own reports, they will have to go through reports of dozens of other companies that have paracetamol in their products.
As shown in Table 1, approximately 23% of organizations or company headquarters had medicinal product(s) containing substances on the pilot list. EMA assuming that companies’ signal management activities are coordinated at headquarters’ or QPPV level, which would mean that 403 and 463 pharmaceutical companies were supposed to be involved in the pilot, but EMA was able to confirm active participation only for around 150 companies based on survey responses and usage data. That suggests that roughly one third of the companies that were legally required to participate in the pilot actually did so.

Table 1 of the Report to EC on signal detection in EV by MAHs
Brexit problems
As stated in our previous blog post, the lack of resources due to Brexit was the original explanation why the pilot was postponed from the end of 2018 until the end of 2019. Brexit also affected other parts of the pilot. Some of the information sharing activities have been scaled down because of EMA’s Brexit Preparedness Business Continuity Plan. Also, the UK was responsible for the highest number of substances (Figure 7) and UK PRAC rapporteurship and signal leadership had to be redistributed to other countries in preparation for Brexit.
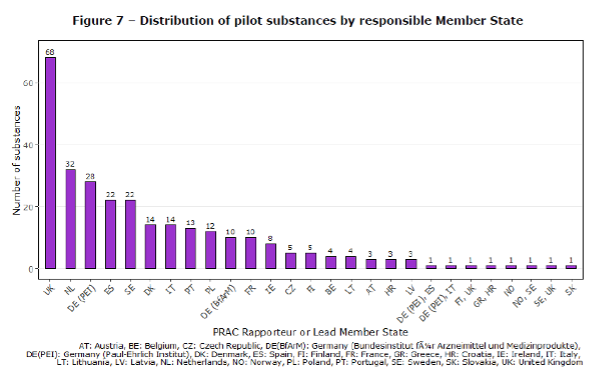
Figure 7 of the Report to EC on signal detection in EV by MAHs
Number of signal notifications
No emerging safety issue originating from EudraVigilance monitoring was received in the context of the pilot during the report period. Six (6) valid signal notifications were received during the first year of the pilot (Table 5).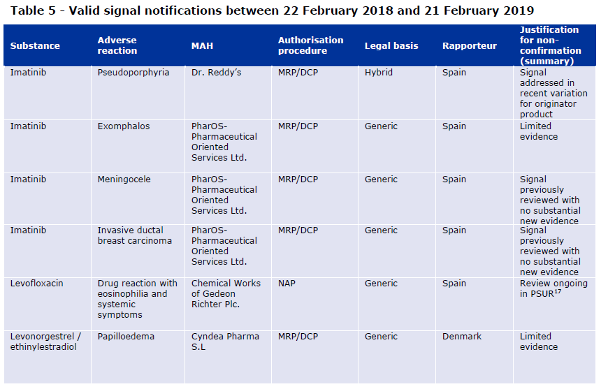
Table 5 of the Report to EC on signal detection in EV by MAHs
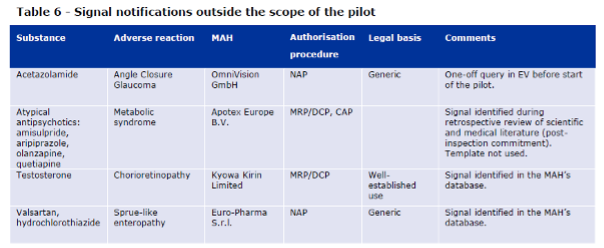
Table 6 of the Report to EC on signal detection in EV by MAHs
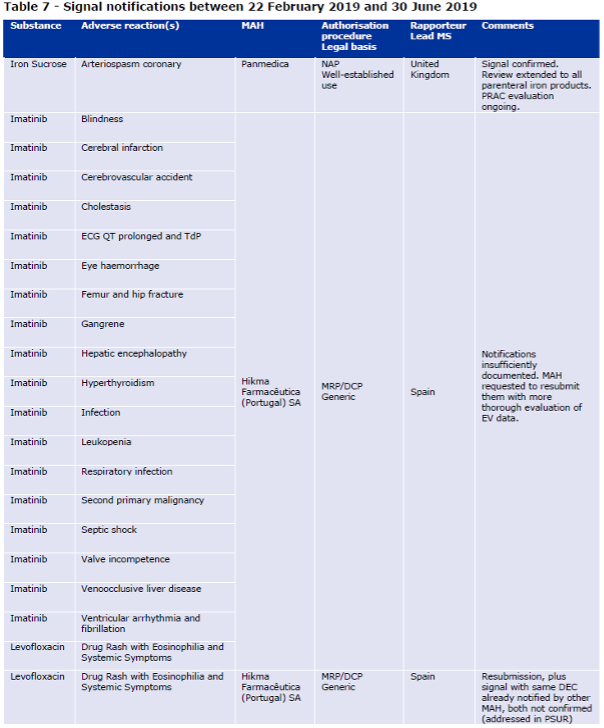
Table 7 of the Report to EC on signal detection in EV by MAHs
Quality of the signal notifications
The signal notification for iron sucrose, the only confirmed signal in the context of the pilot, was well documented according to EMA. The report describes that Panmedica provided a very detailed report, with a clear breakdown of the MedDRA terms used to search EVDAS and details of cases retrieved were provided. According to the agency, the line listing of all cases retrieved from EVDAS (provided as an annex) was very helpful to the assessor when assessing the strength of available evidence. The report states that for this signal, PRAC considered that the available evidence in EudraVigilance and in the literature warranted further evaluation, not only for iron sucrose, but for all parenteral iron products. Relevant MAHs for these products were therefore requested to perform an in-depth cumulative review.
In October 2019, having considered the data from clinical trials and post–marketing use provided by the MAHs, together with the available evidence from literature, PRAC agreed that the strengths of the causal relationship of Kounis syndrome (the concurrence of acute coronary syndromes with conditions associated with mast cell activation) with the use of iron containing medicinal products for intravenous administration is sufficient enough to add a warning to the product information of iron sucrose, ferric carboxymaltose, iron isomaltoside, iron dextran and sodium ferric gluconate containing medicinal products.
The notification for the signal of papilloedema with levonorgestrel/ethinylestradiol and the first notification for the signal of DRESS with levofloxacin were also considered adequately documented by the assessors, although the signals were not confirmed.
The report states that the remaining 23 initial notifications (88%) within the scope of the pilot were generally of poor quality. According to EMA, these notifications focused on quantitative aspects and lacked a clinical qualitative assessment of the evidence. On the topic of clinical qualitative assessment, EMA is referring to guidance in GVP Module IX and in the template for standalone notifications. The group of notifications with poor quality includes the 19 notifications sent on the same day by Hikma. EMA sent them back to the company with a request to perform a thorough evaluation before a resubmission was considered. According to the report, the one signal notification that was resubmitted by Hikma was of better quality.
Duplications efforts
The three notifications that were further processed by the authorities had recently been addressed in regulatory procedures (variation and PSUR). According to EMA’s report, the senders, who were generic companies, were unaware of these regulatory developments. Our guess would be that the “signals” were received from the variation or the PSUR and were notified as signals. According to the report, the same drug-event combination (DEC) (levofloxacin and DRESS) was notified by two different pharmaceutical companies, which represents a duplication rate of 3.8% of all notifications. However, as almost all the signal notifications were not confirmed, it is quite pointless to go into the subject of duplicate reports or whether the other pharmaceutical companies with imatinib or levofloxacin should have sent signal notifications as well. Let’s point out that there are currently 16 other marketing authorization holders for intravenously administered iron sucrose in the Article 57 database and at least 1 company that has ferric carboxymaltose with intravenous administration. In spite of this, Panmedica was the only company that sent the signal notification for arteriospasm coronary. So, either the other companies were unable to detect the signal, analyze it correctly and report, or they did not perform signal detection at all (or were not MAHs during the first year of the pilot).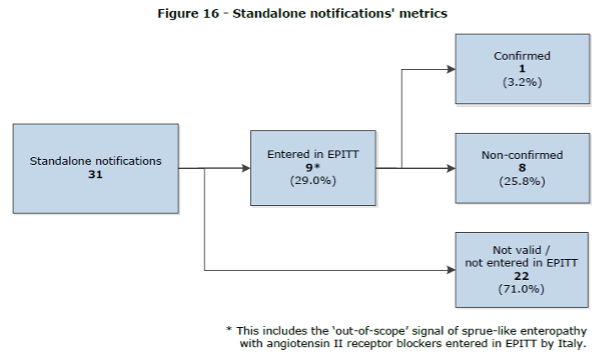
Figure 16 of the Report to EC on signal detection in EV by MAHs
How often companies performed EudraVigilance monitoring during the pilot?
As mentioned above, the pharmaceutical companies, were required to monitor EudraVigilance data using a risk-proportionate frequency, considering the profile of a given active substance or medicinal product. According to the Figure 17 of the report, most companies were performing the detection activities monthly, quarterly or every 6 months. Importantly, the figure does not show the number of the companies but the products. This also means that those number include same substances counted several times. This is one of the parts of the report that shows well how shockingly little data EMA was able to gather during the initial pilot period.
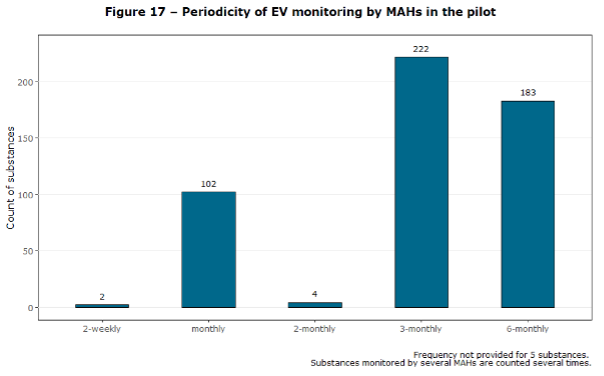
Figure 17 of the Report to EC on signal detection in EV by MAHs
Time effort from the EMA during the pilot
According to the report, EMA dedicated an estimated total of 70 hours to the handling of the 31 standalone notifications received as of June 2019. This includes initial triage, entry into EPITT, tracking, support to PRAC, communication etc. How the time was distributed between valid and invalid signal notifications is detailed in the Table 15. The report describes that between 2 and 6 staff members within the agency’s signal management team were involved in the handling of each notification. The Table 15 shows that the valid confirmed signal notifications require several times more resources than the non-valid (and as described by EMA, badly documented) notifications.
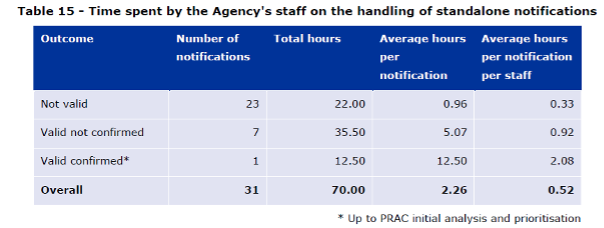
Table 15 of the Report to European Commission on signal detection in EudraVigilance by Marketing Authorization Holders
Table 16 shows the days from confirmation of receipt to the confirmation or non-confirmation. These timelines varied from 9 to 30 days with little to no correlation between the days required and validity of the signal. Admittedly, it would be the best not to talk about correlation with this small amount of data gathered by EMA for the report.
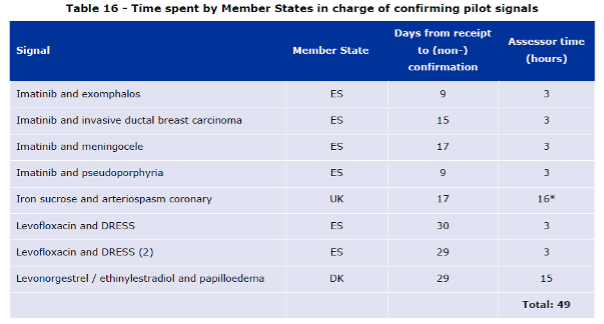
Table 16 of the Report to European Commission on signal detection in EudraVigilance by Marketing Authorization Holders
Estimated time effort required from the EMA if signal detection was scaled
The main question we would want to know the answer to is how much time the MAHs spent on signal detection during the pilot and how much more they would need to spend if the requirement was implemented to all substances. This is not detailed in the report, but the EMA has calculated some other estimates for how much time it would take the authorities to assess the notifications. EMA also collected some survey responses from the pharmaceutical companies involved in the pilot that give a good picture of the time effort required from MAHs.
EMA’s assumption was that substances outside the pilot are more likely to be generics and that there are higher numbers of companies that have the active pharmaceutical ingredient in their products. Most of the standalone signal notifications received during the signal detection pilot concerned generic companies. EMA also assumed that companies outside the pilot may be less experienced in signal detection. Considering these differences and the limited amount of data generated throughout the pilot, EMA admitted that making reliable extrapolations was challenging and the figures they have produced for the estimates should be considered with caution.
EMA used two multiplication factors to make extrapolations: one based on organizations, the second on substances. When the estimations were done, there were 2,138 MAH headquarters in the Article 57 database, 463 of which had a substance in the pilot which corresponds to a multiplication factor of 4.6. According to the EMA, there were approximately 19,655 active substances in EudraVigilance originating from product information in the Article 57 database, hence a multiplication factor of 68.2 from the 288 pilot substances. Also, counts of signals and workload data were brought from 16 months (February 2018 – June 2019) to 12 months, for example the 31 notifications discussed in the report correspond to 23.25 notifications per year.
The report to European Commission states that, based on the figures above, a full implementation of the legal requirements for marketing authorization holders to perform signal detection in EudraVigilance could lead to the notification of 107 – 1,586 standalone signals per year. Of these signal notifications, 28 – 409 signals could be entered in EPITT, and 3.5 – 51 could be confirmed. This would correspond to an average of 0.36 – 4.64 MAH signals per PRAC meeting, considering that there are 11 meetings per year.
From a workload perspective, this would represent 242 – 3,581 hours per year at the Agency, i.e. 4.7 – 68.9 hours per week. For all NCAs, the global impact could be between 169 – 2,506 assessor-hours per year. If workload was spread evenly across all 28 NCAs involved in signal management, this would represent 6 – 89.5 hours for each NCA. Importantly, this only accounts for the confirmation process, i.e. not for further stages of PRAC evaluation.
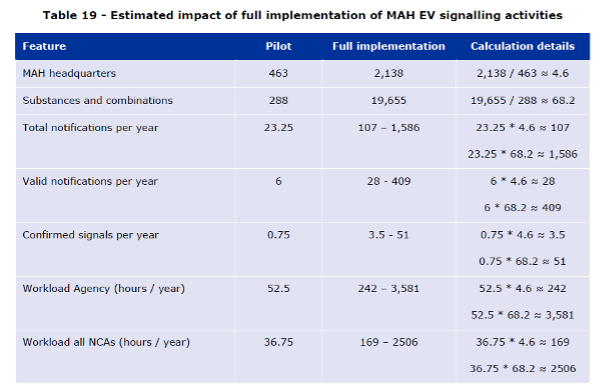
Table 19 of the Report to European Commission on signal detection in EudraVigilance by Marketing Authorization Holders
Additional time effort to the marketing authorization holders
Several companies highlighted in the surveys included to the report that EudraVigilance signaling activities have generated a disproportionate amount of work from their end compared to the outcome in terms of patient safety. According to the survey responses, for new substances EudraVigilance data is almost identical to that available in the corporate safety databases and hence of little added value to the companies, while for old substances there is significant duplication of efforts between generic companies.
The survey to the pharmaceutical companies contains a list of feedback that includes mainly comments about missing detailed information from EMA related to the process, too many duplicate cases, that the amount of work is out of proportion compared to the benefit, about long delays in responses from the EMA, detailed suggestions to improve the data structure of the EVDAS outputs, concerns about the workload if the pilot was extended to all products, etc.

Annex V – Free text comments by respondents to the process survey
For old substances, there is significant duplication of efforts between generic companies. The survey respondents considered EudraVigilance to be a more valuable data source to support the evaluation of signals detected elsewhere rather than it is to detect signals.
Limitations of the pilot
The report contains discussion part with limitations, insights, and conclusion from EMA. We have summarized these here with our own comments.
A potential high volume of standalone signal notifications was one of the concerns that led EMA and the European Commission to agree on a pilot for MAHs to perform signal detection in EV, with an initial focus on a limited number of active substances. The pilot was intended to test and gather experience on the process. However, 16 months into the pilot, a limited number of notifications had been received, from very few MAHs, for very few substances and involving very few rapporteurs.
According to EMA, the choice of the additional monitoring list as a basis for the pilot was driven by several considerations:
- It contains those products which by definition are in greater need of safety monitoring.
- It is an established list with clear inclusion criteria, which avoided having to ‘cherry-pick’ substances based on other criteria that may have been perceived as unfair by concerned MAHs.
- It contains a ‘manageable’ amount of substances and by including all medicinal products containing substances on the additional monitoring list a slightly wider range of situations was covered.
EMA goes on to say that as shown in this report, this choice may have come at the cost of the amount and ability to extrapolate the data gathered through the pilot.
According to the report the exact number of MAHs that effectively monitored EV data during the beginning period of the pilot was unclear. Only 36% of the 463 MAHs that had a substance on the pilot list according to Article 57 data answered the two surveys from EMA, and only 40% of the 463 MAHs made requests in EV for ICSR data level 2B. One of the surveys asked participants to confirm whether or not they participated in the pilot and 26 denied their involvement.
EMA continues that while they have direct access to standalone signal notifications, MAHs have a structured template they need to use and there are good tracking tools in place, they have had problems collecting data on EV signals submitted in PSURs or as variations. The survey responses on these topics did not seem fully reliable after EMA had received more information through follow-ups with the MAHs. During the discussions on the revision of GVP IX, EMA had envisaged to request MAHs to inform them about EudraVigilance signals submitted in PSURs or variations for tracking purposes but this was considered potentially too burdensome.
Another limitation of the pilot was its duration, as one year may not fully cover the entire lifecycle of MAH signaling activities, from getting used to screening EV data to final outcomes. For instance, it was not possible to capture, within the timelines of this report, the final outcome of the PRAC evaluation of the only signal confirmed in the context of the pilot.
Situation update for 2022
As the report covered only the first 16 months of the pilot program, which has been now running for more than 4 years, we contacted EMA to get the latest numbers on signal notifications. What we received was the number of signal notifications reported by the MAHs for 2018-2020. So not exactly the latest figures, but pretty close.
As of December 2020, the Network had received 45 standalone signal notifications from MAHs. Of these, 11 signals were considered valid and processed accordingly, ultimately leading to 1 signal being confirmed for evaluation by the PRAC. These were as follows:
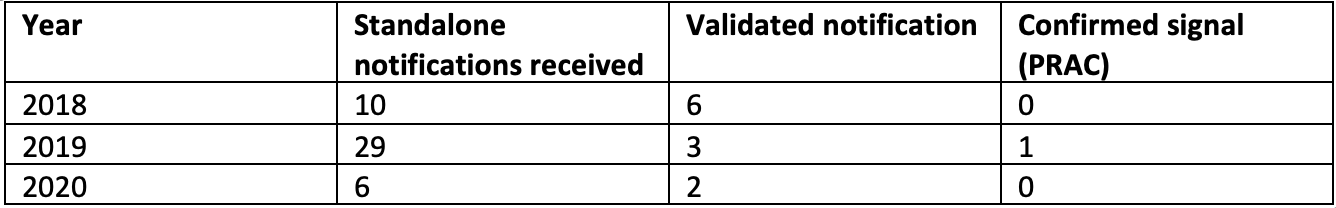
Standalone signal notifications (2018- 2020)
It looks like that the number of notifications has been approximately the same if we exclude the one batch of incorrectly documented signal notifications in 2019 described in the report. Most surprising for us was that the number of the notifications is very low. Based on the huge importance that signal management has in the European pharmacovigilance activities, we would have guessed that there are much larger number of notifications (even if incorrectly documented and not confirmed by the authorities).
Insights of the pilot
The report states that standalone notifications mostly originate from generic companies, but according to EMA, the generic companies are not always aware of recent regulatory outcomes for originator products, which can result in unnecessary notifications and associated workload for both companies and regulators. EMA report stated that including signals in PSURs is not an option for most generic companies since PSURs are not routinely required for their products. “Generic companies can submit variations, but standalone notifications are a more likely route for them.”
During the report period, there was one signal duplication (reported by two different companies). EMA’s assumption was that the duplication of EudraVigilance monitoring activities amongst MAHs with the same substance (e.g. generics) does result in some signals for the same drug-event combination being notified by several MAHs. Duplication in this context might be a bit misleading as there can be several dozens, even hundreds, of companies with the same substance. As EMA states in the report, these ‘duplicate’ signals cannot be simply discarded by the MAHs, nor the regulatory authorities. The companies are legally required to send the notifications to the authorities and the authorities are legally required to enter them in EPITT and considered by the rapporteur in case they contain relevant additional information. EMA highlighted that this is a potential source of unnecessary workload for both the companies and the regulators.
The report states that, based on the quality of evidence provided for the signals, there are differences in the experience of MAHs in signal management. As mentioned before, the report provides that 88% of the signal notifications within the scope of the pilot were of poor quality.
EMA questions the added value for public health of the exercise because a substantial amount of time and resources across involved parties was dedicated to the preparation, running and evaluation of the signal pilot and in spite of all this, only one stand-alone signal was considered worthy of further evaluation by PRAC during the report period.
In addition, EMA points out that substances outside the pilot are generally older, and probably continuous EudraVigilance monitoring by pharmaceutical companies who own products containing these substances would not lead to detection of important signals. It has been analyzed that the median time since authorization for drugs that prompted a signal discussion at PRAC was significantly less (12 years) compared with drugs that had no signal within the same period (20 years).
Conclusion
The EMA report to European Commission states that EudraVigilance signaling activities by marketing authorization holders for substances in the pilot have not been associated with any clear benefit in terms of public health, and it is not expected that a full roll-out of these activities across all EU substances would lead to the identification of many important safety issues.
According to the EMA the exact impact of a full roll-out on workload across all stakeholders (authorities and the pharma companies) is difficult to predict accurately based on the pilot data, but any resources allocated to support such implementation (administrative handling of notifications, evaluation, communication, training, tool development and maintenance, queries, etc.) would effectively be diverted from impactful activities in terms of public health.
EMA suggests that alternative approaches should be considered. Their suggestion was that MAHs would retain access to EudraVigilance as a complementary source of information for their pharmacovigilance processes, but they would no longer be required to continuously monitor the database and nor notify validated signals to the authorities. Such a change would require an amendment to the Commission’s Implementing Regulation on pharmacovigilance.
As we know, it has been almost 3 years since this suggestion was given to the European Commission, but the legislation has not been updated and the companies and authorities involved in the pilot are still required to waste their time in the exercise. In addition, many companies are downloading ICSRs regularly (or paying their service providers for the activity) and performing monthly or quarterly signal detection without an actual requirement to do so.
We hope that European legislators will take action, soon.
And in the meantime, we will continue to focus on our objective to transform pharmacovigilance into a modern, value-based healthcare field. An industry where service providers look after their customers’ best interests rather than making up arbitrary, time-consuming, and expensive activities with no legal justification, such as daily EV downloads or signal management reports.
Did you like the article? Share with your network!
…or tell us your opinion.

Follow our newsletter!
Keep up with industry trends and get interesting reads like this one 1x per month into your inbox.Learn more about Tepsivo
We deliver modern PV solutions to fulfill your regulatory needs using less resources. See how we do it >

0 Comments